L'étude du génome consiste en un repérage des gènes potentiels, dans un premier temps. Ensuite, on cherche à vérifier si le gène putatif à un rôle fonctionnel par comparaison de cette séquence contre des bases de données d'autres séquences connues. C'est l'étape de l'annotation génique qui consiste à déterminer le rôle d'un gène par homologie de séquences. Une autre méthode beaucoup plus directe consiste à utiliser des logiciels de prédiction de gène. Dans ce sens la bioinformatique est un outil puissant pour une annotation rapide des gènes, et un outil qui s'avère nécessaire puisque le nombre de gènes à étudier ne cesse d'augmenter.
Des outils disponibles sur internet ont été mis en place afin de permettre ce type d'étude. Le but de ces travaux pratiques a donc été de nous familiariser avec ces outils, afin de caractériser un gène au sein d'un fragment contigü d'ADN humain.
Les objectifs du TP sont de récupérer ce fragment génomique, d'identifier le gène qui y est contenu, et notamment de mettre en évidence sa structure introns-exons, les différents signaux fonctionnels tels que le promoteur, le site de polyadénylation.... Ceci nous permettra par la suite de déterminer le ou les ARNm issus de la transcription et la séquence protéique correspondante. Ensuite, nous étudierons l'expression de ce gène à savoir les formes alternatives de l'ARNm suite à l'épissage ainsi que sa spécificité tissulaire.
Le protocole de ce TP est basé sur le supplément de Nature genetics de septembre 2002.
1. Récupération du fragment génomique
Nous avons pu récupérer sa séquence nucléotidique grâce au serveur UCSC (http://genome.ucsc.edu) qui contient les séquences génomiques de l'Homme et de la souris.
La séquence étudiée a une taille inférieure à 20 kB, pour des raisons de manipulation de données dans windows. De plus, le gène doit présenter des EST afin de permettre l'étude de son expression, et nous travaillerons sur le brin +.
2. Identification et reconstruction du gène
Une approche entièrement automatique avec l'utilisation d'un programme spécialisé, GENSCAN (http://genes.mit.edu/GENSCAN.html) et une approche par homologie avec l'utilisation de BLAST (http://www.ncbi.nlm.nih.gov/BLAST/).
GENSCAN est un logiciel de prédiction des positions des introns et exons d'un gène. Il est basé sur les modèles de Markov cachés (HMM). On soumet la séquence du contig au logiciel via l'interface web et celui-ci nous donne le résultat sous forme graphique.
L'approche par homologie va nous permettre de déterminer les régions codantes du contig en faisant un alignement de la séquence nucléotidique contre une banque d'EST (BLASTN). Cette première détermination de la position des exons sera affinée par un alignement de la séquence nucléotidique contre une banque non redondante de protéines (BLASTX).
3. Traduction
4. Etude de l'expression
1.Récupération du fragment génomique
La séquence génomique du contig ZNF3 peut être visualisée ici.
2. Identification et reconstruction du gène
2.a. Résultat obtenu grâceà GENSCAN
Les positions des exons déterminées par GENSCAN sont consultables ici.
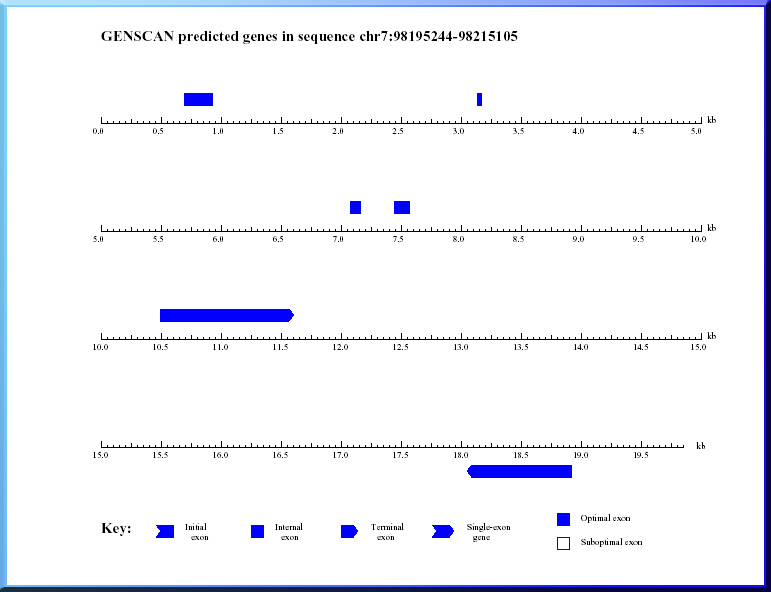
La sortie graphique de GENSCAN pour notre contig est présentée ci-dessous:
Le tableau ci-dessous, présente les différents exons prédits par le logiciel ainsi que leur position sur la séquence soumise.
| EXONS | POSITIONS |
| 1 | 692-928 |
| 2 | 3136-3172 |
| 3 | 7078-7166 |
| 4 | 7447-7573 |
| 5 | 10496-11565 |
GENSCAN a prédit la présence de cinq exons et d'un site de polyadénylation (14323 - 14326). Cependant, on peut remarquer que GENSCAN n'a pas repéré d'exon initial. Il n'a pas non plus repéré de séquence promotrice
2.b. Résultat obtenu par homologie (BLAST)
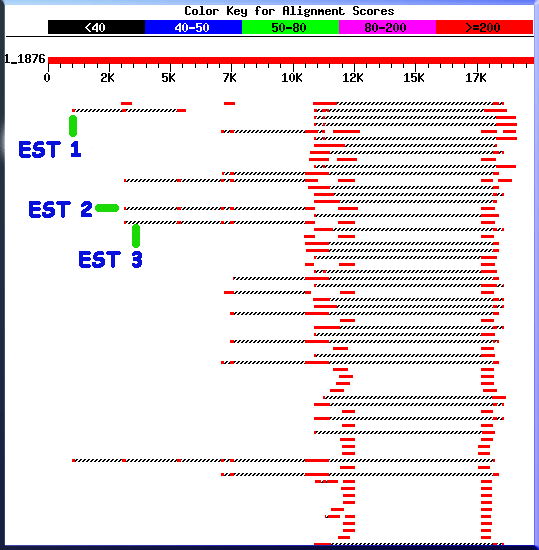
Par contre, les EST 1, 2 et 3 correspondent au critère précédemment défini. Ils ne s'alignent par contre pas avec la totalité de la séquence étudiée.
Les résultats obtenus sont présentés dans le tableau suivant:
| Exon | Position | EST correspondant |
| 1 | 382 - 408 | 1 |
| 2 | 981 - 1073 | 2 et 3 |
| 3 | 3051 - 3168 | 2 et 3 |
| 4 | 3135 - 3172 | 1 |
| 5 | 5272 - 5405 | 1, 2 et 3 |
| 6 | 7077 - 7167 | 2 et 3 |
| 7 | 7444 - 7573 | 2 et 3 |
| 8 | 10495 - 11224 | 2 et 3 |
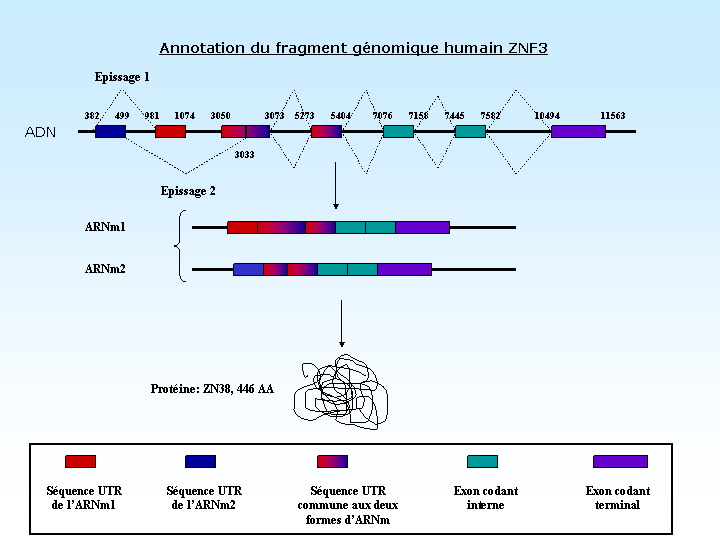
Ci-dessous est présenté le modèle d'épissage alternatif proposé. Nous avons émis l'hypothèse que les deux formes d'ARNm possibles avaient en commun les 4 derniers exons.
 image
image
L'ARNm 1 est constitué des exons 1, 4, 5, 6, 7, 8 (6 exons au total). L'ARNm 2 est constitué des exons 2, 3, 5, 6, 7, 8 (6 exons au total).
La sortie graphique du blast est présentée ci-après:
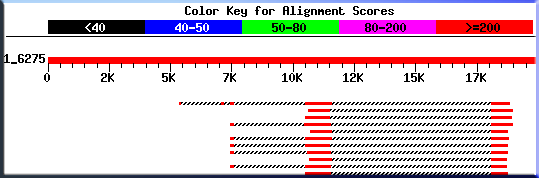
Le tableau ci-dessous présente la comparaison entre les exons précédemment déterminés et ceux présentés sur l'alignement ci-dessus:
| Exon déterminé par blastn | Présence suite au blastx |
| 1 | non |
| 2 | non |
| 3 | non |
| 4 | non |
| 5 | Seulement pour la partie correspondant aux positions 5351 à 5405 |
| 6 | oui |
| 7 | oui |
| 8 | oui |
Nous n'avons pas déterminé d'UTR 3'. Il est possible que nous n'ayons pas réussi à déterminer la fin du gène. Par contre la fin de la séquence alignée issu de la banque nr apparaît bien sur notre séquence. Nous avons donc bien la séquence protéique entière. Celle-ci est présentée ci-dessous :
METQADLVSQEPQALLDS SALPSKVPAFSDKDSLGDEMLAAALLKAKSQ (exon 5) QELVTFEDVAVYFIRKEWKRLEPAQRDLYRDVMLENYGNVFSLGKE (exon 6) DRETRTENDQEISEDTRSHGVLLGRFQKDISQGLKFKEAYEREVSLKRPLGNSPGERLNR (exon 7) KMPDFGQVTVEEKLTPRGERSEKYNDFGNSFTVNSNLISHQRLPVGDRPHKCDECSKSFN RTSDLIQHQRIHTGEKPYECNECGKAFSQSSHLIQHQRIHTGEKPYECSDCGKTFSCSSA LILHRRIHTGEKPYECNECGKTFSWSSTLTHHQRIHTGEKPYACNECGKAFSRSSTLIHH QRIHTGEKPYECNECGKAFSQSSHLYQHQRIHTGEKPYECMECGGKFTYSSGLIQHQRIH TGENPYECSECGKAFRYSSALVRHQRIHTGEKPLNGIGMSKSSLRVTTELNIREST (exon 8)
Détermination de la séquence promotrice : Genscan n'a pas détecté de séquence promotrice.
3. Traduction de la séquence :
Nous avons traduit grâce au logiciel précédemment cité la totalité de la séquence du fragment étudié. La traduction s'est faite dans les trois cadres de lecture possibles et nous a permis d'affiner la position des exons, en déterminant les jonctions intron/exons et le codon-stop.
Les jonctions intron/exons sont caractérisées par la présence d'un site donneur et d'un site accepteur. A la fin d'un exon on doit retrouver un dinucléotide AG, et au début d'un intron on doit retrouver un dinucléotide GT. Ce sont ces dinucléotides que nous avons recherchés afin de déterminer la position exacte des exons sur notre séquence.
Le tableau ci-dessous présente les résultats obtenus, comparés à ceux déterminés précédemment :
| Exon | Position d'après Blast | Position d'après la recherche des jonctions intron/exon |
| 1 | 382-408 | 382-499 |
| 2 | 981-1073 | 981-1074 |
| 3 | 3051-3168 | 3050-3073 |
| 4 | 3135-3172 | 3050-3073 |
| 5 | 5272-5405 | 5273-5404 |
| 6 | 7077-7167 | 7076-7158 |
| 7 | 7444-7573 | 7445-7582 |
| 8 | 10495-11224 | 10494-11563 |
Nous avons recherché des informations sur le gène ZNF3 sur le site d'Ensembl (http://www.ensembl.org/Homo_sapiens/geneview?gene=ENSG00000166526). C'est un logiciel de prédiction de transcription et de traduction basé entre autres sur Genscan ou Genewise. L'information sur ce site prévoit la traduction du gène en trois protéines, toutes étant des protéines à doigts de zinc de 392, 131 et 446 acides aminés respectivement. Cette dernière correspond à la protéine que nous avons déterminée par homologie. Les renseignements collectés sont les suivants :
protéine HZF3.1 ayant un domaine KRAB, protéine à doigts de zinc 38 impliquée dans la différenciation cellulaire et/ou la prolifération cellulaire. Elle serait localisée au niveau du noyau cellulaire, sans spécificité tissulaire.
La séquence protéique a déjà été précédemment déterminée grâce au blastx. Nous nous sommes servis de celle-ci pour déterminer les cadres de lectures dans lesquels se faisait la traduction de chaque exon, par rapport au début de la séquence. Le tableau ci-dessous présente les résultats correspondant :
| Exon | Cadre de lecture de la traduction |
| 5 | 2ème |
| 6 | 3ème |
| 7 | 1er |
| 8 | 1er |
4. Etude de l'expression
Nous avons regardé l'origine tissulaire (fiches GenBank) des EST alignés sur notre séquence par BLASTN. Voici quelques organes où sont exprimés ces EST :
utérus, coeur, foie,rate, rein, estomac, rétine, cerveau (hypothalamus), prostate, poumon, placenta...
Nous n'avons pas observé de prédominance d'un organe particulier, donc on en conclut qu'il n'y a pas de spécificité tissulaire.
Nous avons étudié un contig humain selon deux méthodes. La première basée sur le logiciel de prédiction Genscan nous a fourni une annotation automatique de cinq exons contenus dans le contig, ainsi que d'un site de polyadénylation. La deuxième basée sur l'homologie de séquences a conduit à l'annotation de huit exons, dont les quatre premiers ne sont pas traduits. En effet, nous avons détecté un épissage alternatif conduisant à deux formes d'ARNm, ne différant seulement qu'au niveau de la région non traduite. Ainsi, la traduction de ces deux ARNm conduit à la synthèse de la même protéine. Cependant, cette étude reste incomplète puisque nous n'avons pas pu mettre en évidence le site promoteur, l'UTR 3' et le site de polyadénylation.
Cependant, en recherchant des données sur notre contig sur Ensembl, nous nous sommes aperçus de la divergence de nos résultats. En effet, Ensembl prédit la synthèse de trois protéines différentes à partir du contig, dont celle que nous avons déterminée grâce au BLASTX (100% d'identité). Si on prend Ensembl comme référence, il nous manquerait deux protéines, correspondant à un épissage alternatif supplémentaire dans la région codante. On a aussi pu voir la limite du logiciel Genscan.